On
appelle mécanisme réactionnel l'ensemble supposé
des étapes élémentaires justifiant la
transformation observée expérimentalement. Le mot
"supposé" n'est pas anodin...En effet , la réalité
des phénomènes ne sera jamais vraiment
élucidée.
La
représentation d'un mécanisme
est relative à un modèle choisi. Si
le modèle est bon, en plus de justifier les résultats
expérimentaux, il permettra au mieux
de prévoir un résultat. A défaut, il permettra
d'influencer une recherche dans un but
précis...Le mécanisme représente donc la
réalité relative au modèle choisi.
Différentes modélisations existent :
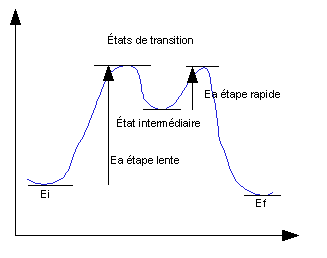
Le
schéma Energie / Coordonnée de réaction
traduit les variations relatives d'énergie du système au
cours de la réaction. Il permet de définir l' (les)
étape(s) lente(s). Ces schémas permettent de distinguer
les états d'activation, hypothétiques, correspondant
à un maximum d'énergie, des états
intermédiaires, isolables ou à défaut,
observables, qui sont des minimums d'énergie. Ils permettent
aussi de visualiser quelles sont les étapes lentes
(d'énergie d'activation élevée) et les
étapes rapides (d'énergie d'activation faible).
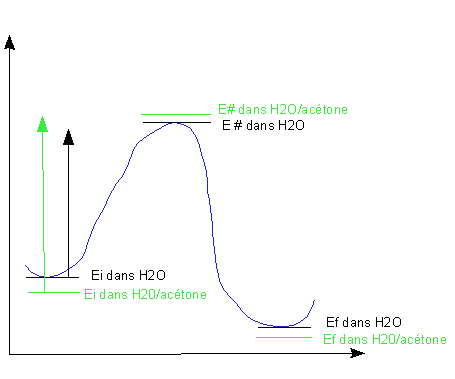
 L'influence de
la
modification de certains paramètres
L'influence de
la
modification de certains paramètres s'étudie de
façon élégante par ce genre de diagramme.
Par exemple l'influence du choix du solvant sur une SN
1
ou
sur une SN
2 peut facilement se montrer:
Soit la SN du
1-chloro propane par l'iodure I
-
. Cette
réaction est a priori d'ordre 2 dans la mesure où le
dérivé halogéné de départ est
primaire.
Bilan: CH
3-CH
2-CH
2-Cl
+ I
-
--> CH
3-CH
2-CH
2-I
+ Cl
-
Etudions cette réaction supposée d'ordre 2 dans deux
solvants différents , l'eau et un mélange
eau/acétone. Une réflexion nous incite à penser
que l'état initial sera plus stable dans ce solvant: en effet,
RCl est plus soluble (cad mieux solvaté, cad plus stable) dans
l'acétone , solvant organique,que dans l'eau pure. I
-
quant à lui sera mieux solvaté dans l'eau pure mais il
reste bien solvaté dans ce mélange.
La notion de
contrôle cinétique, opposé au contrôle
thermodynamique, est le résultat de l'analyse de
résultats expérimentaux à l'aide de ce genre de
diagrammes.
Analysons un
processus sous contrôle thermodynamique:
Par exemple,
l'élimination
de HBr à partir d'un
dérivé halogéné, conduit
expérimentalement systématiquement à
l'alcène le plus stable, indépendamment de
l'acidité du H arraché. Comparons les schéma E/CR
selon les deux processus possibles dans le cas d'une
élimination de type E2 sur le 2-bromo 3-méthyl butane.
Deux alcènes sont envisageables.
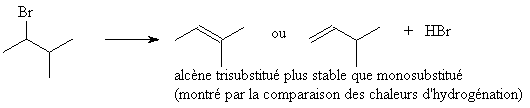
Ces alcènes sont obtenus via la rupture de deux liaisons C-H,
dont la force peut être comparée grâce à
l'étude des couples A/B correspondants:
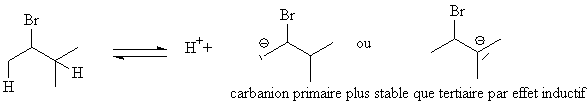
L'hydrogène qui donne
rait le carbanion le
moins stable est a
priori lié par une liaison plus forte que l'autre, mais conduit
à l'alcène le plus stable.
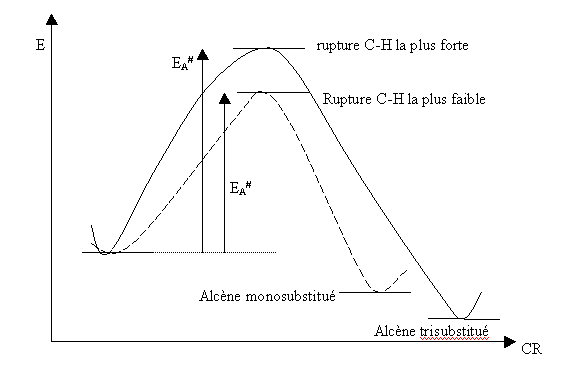 L'expérience
L'expérience montre
que le chemin réactionnel donnant
l'alcène le plus stable, malgrè la plus grande
énergie nécessaire pour arracher l'H, est largement
prioritaire.
C'est
donc le gain d'énergie la plus grand entre l'état initial
et l'état final qui décide du produit de la
réaction. C'est donc un raisonnement d'ordre thermodynamique qui
permet de prévoir le résultat de la réaction: la
réaction est dite sous contrôle thermodynamique.
La réaction se produit majoritairement par une étape de
grande énergie d'activation, malgrè la possibilité
d'une voie plus rapide. Les considérations cinétiques
sont sans intérêt pour une réaction sous
contrôle thermodynamique. Toutefois, on conçoit que
la
voie thermodynamique nécessite une température minimale
pour franchir la barrière d'activation élevée.
Toutes
les réactions d'élimination du
programme
sont ainsi sous contrôle thermodynamique. Les éliminations
de HX ou de H
2O,
quelles que soient les conditions expérimentales, se produisent
sous contrôle thermodynamique, c'est à dire donnant
l'alcène le plus stable.
Pour prévoir l'alcène
obtenu, on compare les stabilités des alcènes possibles .
Un
alcène est d'autant plus stable que :
1- Il est aromatique
(mais ce n'est même plus un alcène...)
2- Il est stabilisé
par résonance, avec une longue chaîne de délocalisation
3- Il
porte des groupes alkyl (on le dit substitué)
Analysons un processus
sous contrôle cinétique: L'addition
électrophile
de HBr sur un alcène disymétrique conduit toujours au
dérivé bromé tel que le brome est fixé sur
le carbone fournissant le carbocation le plus stable:
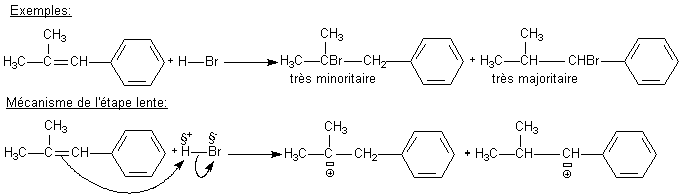
énergie E1
énergie E2
Le
carbocation en alpha du benzène est stabilisé par résonance et donc E2
< E1
Un
schéma énergie / coordonnées de réaction
permet alors de visualiser l'aspect énergétique du
"choix" réactionnel.
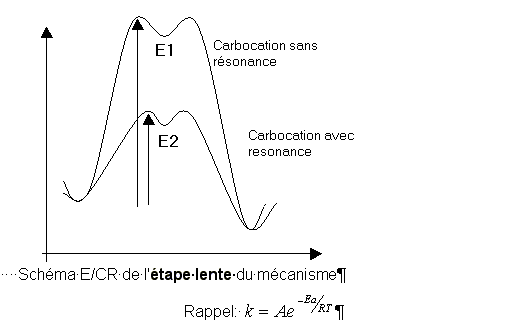
L'énergie
d'activation qui conduit au carbocation d'énergie E2 est donc
plus faible que l'énergie d'activation qui conduit au
carbocation d'énergie E1.
Le carbocation stabilisé
par résonnance est donc obtenu plus vite que le
carbocation ne présentant pas de résonance.
L'expérience
montre que le dérivé bromé majoritaire est celui
issu du carbocation le plus stable, et ce systématiquement
quelquesoit le choix. La réaction obéit donc à un
choix cinétique, elle est dite sous contrôle
cinétique.
De
nombreuses réactions du cours sont sous contrôle
cinétique : l'addition de HX, radicalaire ou
ionique est sous
contrôle cinétique.
Les polymérisations
radicalaires ou anioniques sont sous contrôle cinétique.
les substitutions électrophiles
sur le benzène ou sur les
dérivés aromatiques sont sous contrôle
cinétique (étude des intermédiaires de Wheland)
la réaction de Diels Alder
est sous contrôle cinétique
Pour
prévoir le produit de réaction via des
intermédiaires radicalaires ou via des carbocations sous
contrôle cinétique, on compare donc les énergies
des radicaux ou carbocations formés dans l'étape lente pour choisir le
plus stable.
Un radical ou un
carbocation est d'autant plus stable que , dans l'ordre :
1- il est stabilisé
par résonance . Plus la chaîne de délocalisation est longue, plus il
est stable.
2-
Il porte de groupes donneurs d'électrons par effet inductif.
Les
groupes alkyl ont un effet donneur d'électron, stabilisant ; un
groupe CN ou C=O est un groupe attracteur d'électrons,
déstabilisant.
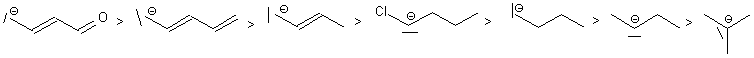
On
remarquera la particularité des nitriles conjugués
capables de fournir des carbanions stables en alpha de CN, au
même titre que un carbanion est stable en alpha de C=O:
La
réaction de Diels Alder est aussi sous
contrôle cinétique mais c'est le modèle de
Hückel qui permet de prévoir quel est l'état de
transition le plus bas (car la réaction est en une étape)
.
On
peut aussi représenter les mécanismes en se focalisant
sur les changements de structure électronique, le ruptures ou
formations de liaisons. Deux modèles existent.
Le modèle de Lewis appliqué
à la réaction chimique tente de représenter les
mouvements
électroniques, obéissant à des principes
électrostatiques, ou de recherche de minimum d'énergie,
dans la formation de la liaison la plus stable.
Les
électrons sont associés en doublets,
représentés par un trait . Un électron
célibataire est représenté par un point.
Une
transformation chimique traduite par un bilan de réaction se
produit par une succession de transformations
élémentaires, passant soit par des états
d'énergie maximum (états de transition) que le
modèle de Lewis ne tente pas de représenter (voir
schéma E/CR) , soit par des entités de faibles
durée de vie que sont les espèces intermédiaires,
que le modèle de Lewis représente.
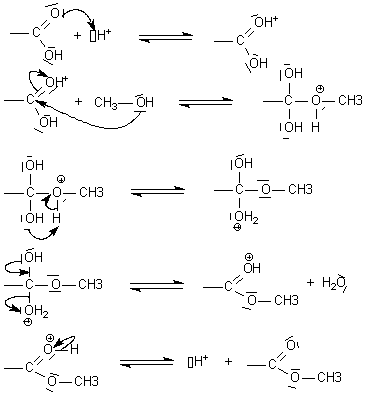
Exemple
:
Bilan de
l'estérification de l'acide acétique par le méthanol en présence de H+:
(H
+)
CH
3COOH
+ CH
3OH
= CH
3COOCH
3
+ H
2O
Mécanisme
dans un formalisme de type étude cinétique : plusieurs étapes
élémentaires :CH
3COOH +
H
+
= CH
3COOH
2+
.
CH
3COOH
2+
+ CH
3OH
= CH
3COH(CH
3OH
) OH
+ CH
3COH(CH
3OH
) OH
+ =
CH
3CO(CH
3OH
) OH
2+ CH
3CO(CH
3OH
) OH
2+
= CH
3CO(CH
3OH
)
+ +
H
2O
CH
3CO(CH
3OH
)
+ =
CH
3COOCH
3
+ H
+ Chaque
étape élémentaire écrite représente
un bilan réellement "vécu" par le système en
réaction . Les techniques modernes ont permis de les observer,
malgrè leur faible durée de vie. En plus , le
modèle de Lewis propose pour ces étapes
élémentaires de justifier des transformations dans les
répartitions électroniques, par des flèches
très codifiées:
Une flèche
représente un mouvement électronique : elle part TOUJOURS
d'un doublet d'électrons vers un centre d'accueil dit
électrophile (ou acide s'il s'agit d'un H).
Le doublet
d'électron au départ
de la flèche est un doublet dit nucléophile (ou basique s'il se dirige
vers un H). On dit qu'il est porté
par une entité nucléophile (ou basique s'il attaque un H).
Peu
importe qu'il soit un
doublet libre ou un doublet lié, de type sigma ou de type pi...
Au
cours de ces mouvements électroniques, les règles de
consruction des atomes doivent absolument être respectées
, en particulier la règle de l'octet . Les
éléments de la première et deuxième
période de la classification périodique ne supportent pas
de dépassement de la règle de l'octet . Un déficit
par rapport à la règle de l'octet est forcément un
centre électrophile (parmi d'autres) et est indiqué
obligatoirement par un rectangle signifiant une lacune
électronique (associé ou pas à une charge +, peu
importe...)
Ainsi,
même un mécanisme en une seule étape peut
être développé selon le modèle de Lewis :
La SN
2
, la décarboxylation ou la réaction de Diels Alder en sont de bons
exemples.
Classement des
mécanismes Les mécanismes peuvent alors
être classés, en fonction de leur
bilan
d'une part et du
type
d'action intervenant dans l'étape lente .
Le
premier mot dans le nom d'un mécanisme est relatif au bilan de la
réaction étudiée.
-On
rencontre donc SUBSTITUTION
;
ADDITION ;
ELIMINATION
Le
deuxième mot s'il existe caractérise le type d'attaque
sur l'entité organique de référence dans
l'étape lente.
-On
rencontre donc NUCLEOPHILE ELECTROPHILE
RADICALAIRE
CONCERTE
Exemple
: H
2C=CH
2 +
HBr = H
3C=CH
2Br
Bilan :
addition
(de HBr sur l'entité de référence alcène)
Mécanisme:
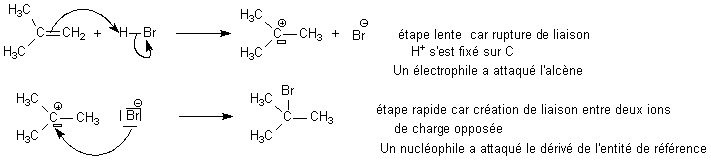
L'étape lente
consiste en l'action de H+ regardé comme un
électrophile
.
Le
mécanisme sera donc une
addition
électrophile (ce qui
signifie bilan= addition , dans l'étape lente = action
d'un électrophile sur l'entité de référence)
Autre
exemple: Ph + E+
= Ph-E +
H+
Bilan :
substitution
d'un H du benzène par E
Mécanisme:
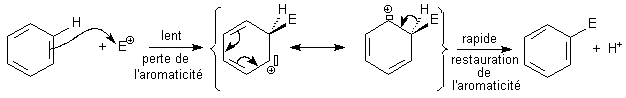
action de l'électrophile E+
élimination de H+
L'étape lente consiste en l'action de E+
électrophileLe
mécanisme sera donc une
substitution
électrophile (ce qui signifie bilan =
substitution, dans l'étape lente = action d'un électrophile )
Autre exemple :
Ph-(C=O)-CH
3 +
R- + EtOH = Ph-C(OH)(CH
3)-R
+ EtO-
Bilan :
addition
de R et H sur la cétone
Mécanisme :
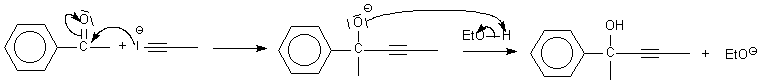
étape lente
car rupture de liaison
étape rapide A/B
action du nucléophile R-
L'étape
lente consiste en l'action du
nucléophile
R-
Le mécanisme sera donc une
addition nucléophile
sur un carbonyle ( ce qui siginifie bilan = addition, étape lente =
action d'un nucléophile )
Le terme
concerté
est relatif à des mécanismes où toutes les
transformations électroniques se font dans le même temps
de sorte qu'il soit difficile de nommer un électrophile, un
nucléophile ou même un radical. C'est le cas pour la
réaction de Diels Alder:
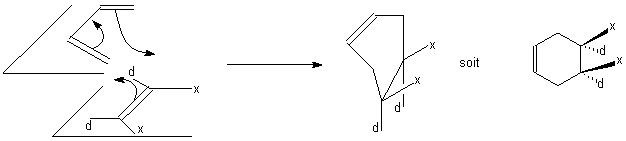
Où
ni le diène ni le diénophile ne sont identifiés
comme électrophile ou nucléophile selon le modèle
de Lewis : c'est un mécanisme d'
addition
concerté en une étape.
D'autre
dénominations existent :
réactions
de type A/B dans
laquelle un atome gagne ou perd un proton via un
doublet
libre ,
sans rupture de
liaison chimique autre que celle dans laquelle
l'H est engagé.
Ainsi la première
étape
de l'addition électrophile de HBr sur un alcène n'est pas
une réaction A/B car la liaison pi C=C a été
rompue
(voir) . Mais la deuxième étape
de l'addition
nucléophile du nitrile sur la cétone dans le solvant
éthanol est typiquement de type A/B
(voir) Il
arrive qu'un mécanisme possède
plusieurs
étapes lentes...on les nomme alors les deux: c'est le cas
du mécanisme de
l'estérification
dans lequel la deuxième étape d'addition
nucléophile est lente alors que la quatrième étape
d'élimination de H2O est lente aussi, alors que le bilan est
celui d'une substitution...On nomme ce mécanime de type
"addition élimination"
plutôt que substitution...pour signaler les deux étapes
lentes...et on ne précise pas nucléophile relative
à la première étape lente...délicat, je
vous l'accorde...
Les réactions d'oxydoréduction
par des espèces minérales ne sont pas
représentées en Lewis , sauf les réductions par
les borohydrures et les alumino hydrures :
Sur les esters ce sont
des mécanismes d'addition élimination alors que sur les
aldéhydes ce sont des mécanismes d'addition (bilan
d'addition de H
2) nucléophile (action du
nucléophile H
- dans l'étape lente) :
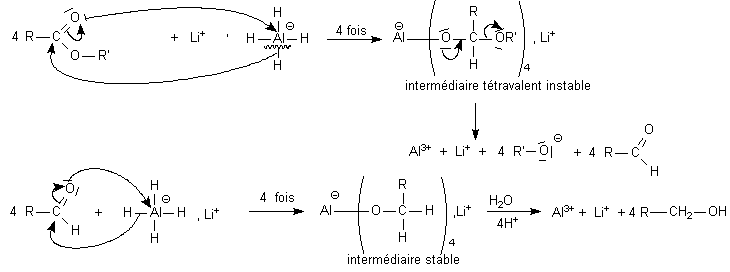
Le modèle des orbitales
frontières de Hückel
utilise les orbitales moléculaires et recherche la combinaison
d'OM favorable en terme d'énergie en réfléchissant uniquement sur les
orbitales frontières.
C'est ainsi que la formation
des liaisons lors de la réaction de Diels Alder sera représentée ainsi
:
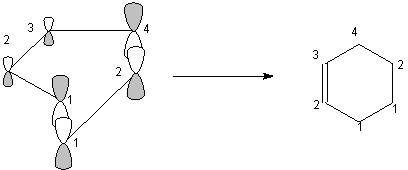
Le
recouvrement entre lobes de même signe deux à deux (ou de
même couleur deux à deux ) traduit la formation
d'une liaison sigma si le recouvrement est dans l'axe de la liaison ou
formation d'une liaison pi si le recouvrement a lieu de part et d'autre
de l'axe de la liaison.
Seules les réactions sous
contrôle cinétique peuvent être
modélisées de cette façon.
La prévision de la
réaction provient des règles suivantes : Contrôle orbitalaire:Après
avoir trouvé les OF de chaque réactif :
-on cherche le couple (HO / BV) des deux
réactifs les plus proches en énergie car plus les
énergies sont proches et meilleur est le recouvrement.
- On vérifie, si plusieurs sites d'une
même molécule réagissent en même temps, que
la réaction est permise par symétrie , c'est à
dire que tous les recouvrements souhaités sont constructifs
(entre lobes de même signe deux à deux )
- On cherche alors le + gros coefficient
en valeur
absolue de la HO et de la BV...ce sont les sites réactifs
prioritaires qui imposent la régiosélectivité de
la réaction.
Si l'expérience est conforme
à ces règles, on en déduit que la réaction
est sous contrôle orbitalaire.
La
réaction de Diels Alder est sous contrôle orbitalaire.
L'addition
d'un cuprate lithié sur une alpha-énone est sous
contrôle orbitalaire
(attaque du nucléophile sur le carbone en position 4 car porteur
du + gros coeff de la BV, position par rapport à l'O
numéro 1), soit une addition 1-4 .
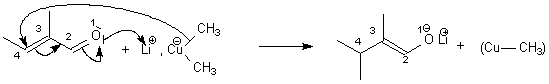
Après hydrolyse et tautomérie
cétoénolique, on obtient un aldéhyde.
Contrôle de charge :Si
l'expérience donne un résultat NON conforme au
contrôle orbitalaire, on peut chercher un contrôle de
charge : la réaction serait-elle contrôlée par des
données électrostatiques ? On cherche donc , pour la
molécule ayant la BV active, l'atome portant la plus grande
charge +, alors que pour la molécule intervenant par sa HO, on
cherche l'atome ayant la plus grande charge - , le tout grâce
à la formule :
Le
calcul ainsi fait donne la charge en électron, il faut
retrancher cette valeur au nombre de protons du noyau relatif aux
électrons pz engagés dans la résonance (soit 1
pour C ou tout hétéroatome à 1 électron cad
doublement lié et 2 pour tout hétéroatome à
2 électrons cad simplement lié) .
Toute
réaction pouvant être justifiée par l'action de
l'atome de charge + la plus forte sur l'atome de charge - la plus forte
de l'autre molécule réactante sera dite sous
contrôle de charge.
L'action d'un organolithié sur
une alpha-énone est sous contrôle de charge (attaque du
nucléophile sur le carbone en position 2, car porteur de la +
grosse charge+, position par rapport à l'O en position 1 ), soit
une addition 1-2.
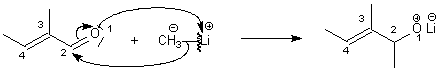
Après hydrolyse
, on obtient un alcool.
L'aspect stéréochimique des
réactions
, c'est-à-dire leur étude dans un espace à 3
dimensions, prend une grande importance, particulièrement pour
la synthèse de molécules médicamenteuse. La
nature, grâce aux enzymes est capable de
stéréosélectivité extrêmement
précise que le chimiste tente laborieusement de reproduire...
En
règle générale, c'est le
caractère
CONCERTE
des réactions qui leur donne une propriété
stéréochimique, que ce soit de la
stéréosélectivité ou de la
stéréospécificité. Toutefois, des effets
stéréochimiques, peuvent donner à des
réactions non concertées, une propriété de
stéréosélectivité.
Une réaction est
stéréosélective,
si , quelle que soit la caractéristique
stéréochimique du réactant, le produit obtenu
présente une propriété
stéréochimique unique.
La réaction typiquement
stéréosélective de votre cours de chimie organique est l'addition de H
2
sur un alcyne, en présence d'un catalyseur solide empoisonné :
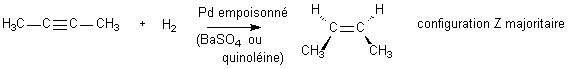
L'alcyne
de départ ne présente aucune stéréochimie .
Alors que l'alcène obtenu peut exister sous une
configuration Z ou E, seule , (ou du moins très majoritairement)
, la configuration Z est obtenue.
Une autre réaction
partiellement stéréosélective peut être l'
élimination de type E
1 sur un dérivé
halogéné :
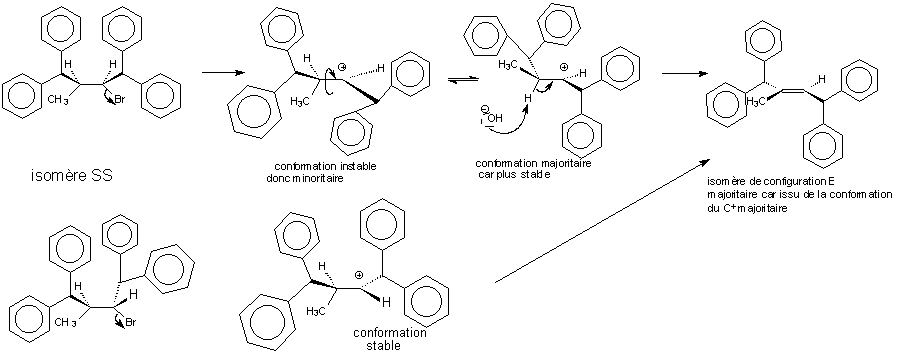
isomère SR
Quelquesoit
l'isomère de départ, l'alcène
majoritairement obtenu est l'alcène E présentant la
moindre gêne stérique. La réaction est bien
stéréosélective.
La réaction est
stéréospécifique,
si , dans la mesure où le réactant présente
plusieurs formes stéréochimiques, la
stéréochimie du produit obtenu est dépendant de la
forme stéréochimique du réactant.
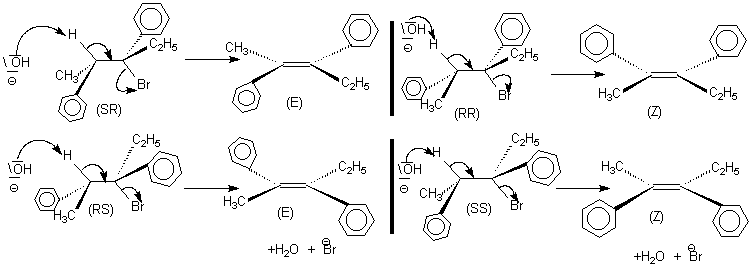
Les réactions
typiquement stéréospécifiques sont la SN
2 (dite
énantiospécifique), la E
2 ,
l'hydrogénation des alcènes , l'addition de Br
2 sur
les alcènes , l'époxydation des alcènes,
l'hydrolyse basique des époxydes, la dihydroxylation d'un
alcène par le tétraoxyde d'osmium (toutes
diastéréospécifiques), la réaction de Diels
Alder.
On remarquera que toutes ces réactions sont
concertées :
Stéréochimie de la SN2
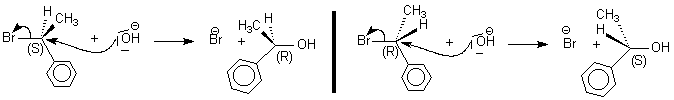
Stéréochimie
de la E2
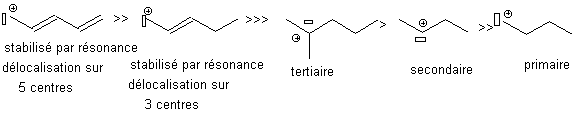

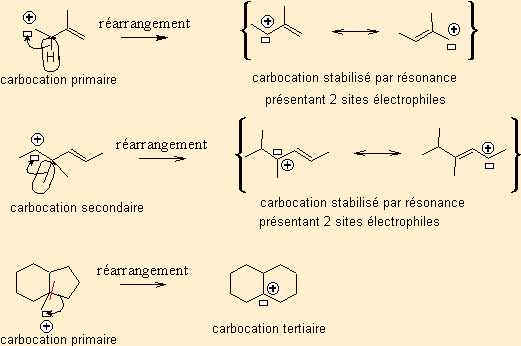 Une liaison σ C-H se rompt . -H migre sur le C+ . Le nouveau carbocation est beaucoup plus stable, car conjugué.
Une liaison σ C-H se rompt . -H migre sur le C+ . Le nouveau carbocation est beaucoup plus stable, car conjugué.