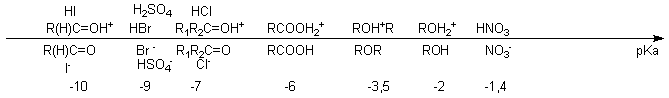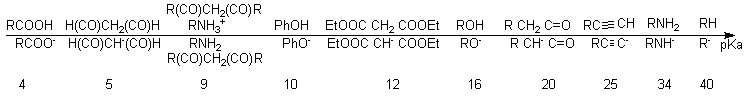
On
rappelle ici que
la force d'un acide est liée à la
stabilité de sa base conjuguée : pour comparer
deux
acides, on compare les stabilités des bases conjuguées et
donc en particulier si possible la délocalisation de la charge.
Plus une charge est délocalisée, plus l'espèce
chargée est stable, et plus l'acide associé est fort.
On
peut donc
arracher un H acide à un produit organique de
façon quantitative avec une base de pKa supérieur.
Toutefois, une réaction A/B peu avancée pourra en chimie
organique produire une quantité suffisante de réactif
instable, donc très vite consommé, ce qui
déplacera la réaction A/B qui finit par devenir
quantitative.
Pour
obtenir les bases les plus fortes, on est obligé d'utiliser des
méthodes redox faisant intervenir des métaux
réducteurs, produisant H
2 à partir du proton
arraché.
On rappelle ici quelques exemples de votre cours de chimie
organique
mettant en évidence les propos ci-dessus. vous pourrez pointer
directement sur ces exemples par les liens ci-contre, à gauche.
Obtention
de la base associée d'un alcyne vrai: on a
les
deux solutions:
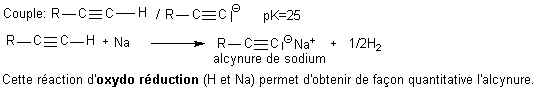
ou
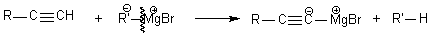
La réaction est totale car
l'organomagnésien est
assimilable à R'
- , base conjuguée de RH, dans
un couple de pKa = 40
ou
R - C
≡ C - H + NH
2- → R - C
≡ Cl
- + NH
3 (en milieu anhydre ) , réaction totale avec toute base de pKa >25
(méthode peu courante...)
Les alcoolates
On peut les obtenir par l'action du sodium comme ci-dessus
avec
l'alcyne vrai.
R-O-H +
Na
-> RO
-
, Na
+
+ 1/2 H
2
alcoolate de sodium
Ils détruisent les organomagnésiens comme les alcynes
vrai ( mais cette méthode trop coûteuse n'est pas utile en
synthèse ) .
R-O-H +
R'-Mg-Br
->
R-O-MgBr + R'-H
Réaction
A/B totale
K= 10
22
Ce sel de magnésium n'est pas utile
car il
neprésente pas de propriété nucléophile
intéressante . L'action de l'eau sur ce sel de magnésium
,
en pH acide pour
éviter la
précipitation de Mg(OH)2
redonne l'alcool par
une réaction A/B à nouveau totale , c'est l'hydrolyse
:
R-O-MgBr
+ H
+
--> R-O-H
+ Mg
2+
+ Br
-
Réaction A/B totale K=10
18
On préfère obtenir les alcoolates à faible
coût par dissolution de potasse KOH dans l'alcool pur .
La solution est de la potasse
alcoolique.
Compte tenu des pKa , la réaction souhaitée pour obtenir
l'alcoolate présente une contante faible:
OH
- +
RO-H <=>
H
2O
+ RO
-
K= 10
-18 / 10
-14
= 10
-4
Mais, même avec un avancement faible, cet équilibre
est
déplacé par la consommation de RO
-
dans une réaction chimique se déroulant dans le milieu,
par exemple une
réaction
de
Williamson :
RO
- +
R'-Br
->
R-O-R'
+ Br
-
Dans
la réacton de Wittig
il faut arracher un H à un sel de phosphonium, pour obtenir
l'ylure de phosphore. Le couple phosphonium / ylure présente un
pKa très élevé de sorte que seules des bases de
type R-NH
- ou R
-
conviendront. Il
faut adapter le solvant à de telles bases. Il doit
être bien sûr aprotique, ou donner une base
conjuguée aussi active que la base introduite.
Ainsi dans la réaction de Wittig une base courante est le n
butyl lithium ou Bu
- Li
+
dans le DMSO ou
diméthyl sulfoxyde ou (CH
3 )
2
S=O .
En réalité , le pKa de Bu
- est
suffisant pour arracher un H au DMSO par une réaction quasi
quantitative:
Bu
- +
(CH
3 )
2
S=O
<=>
BuH + (CH
2
)
-
(CH
3 ) S=O et c'est cette
base DMSO
-
qui est la base active capable d'arracher le proton de l'ylure de
phospsore.
Pour
exploiter les propriétés nucléophiles des amines
, on se place communément en présence d'une base faible,
mais néanmoins légèrement plus forte que l'amine
nucléophile , et non nucléophile elle-même, en
effet, si nous analysons le mécanisme:
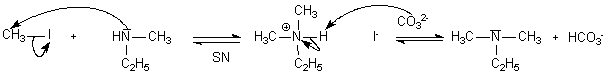
base (10)
acide (10)
base(10,2)
base(10)
ampholyte
(6.3 et 10,2)
Entre parenthèses on trouve les pKa des espèces en jeu.
La base utilisée ici (carbonate) , non nucléophile par
rapport aux dérivés halogénés, est
proposée en excès. C'est la base la plus forte du milieu,
en plus grande quantité, et elle consommera les protons de
l'acide formé à l'issue de la SN. L'amine
nucléophile n'est donc jamais engagée de façon
totale dans un équilibre A/B qui supprimerait ses
propriétés nucléophiles.
On utilise aussi couramment,à la place d'un carbonate, la
pyridine ou une amine tertiaire , toutes les deux non
nucléophiles et légèrement plus basiques.
La
situation est
plus délicate lorsqu'une catalyse acide est nécessaire,
alors que l'on souhaite exploiter les propriétés
nucléophiles des amines. C'est le cas pour l'addition d'une
amine sur un carbonyle pour donner une imine substituée:
H+
+ R-NH2
<=> R-NH3+
pKa = 9,2
H+ + C=O
<=> C=OH+ pKa
» -8
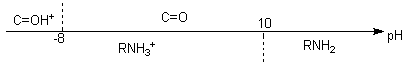
Les
espèces actives dans les
mécanismes envisagés sont R-NH2 et C=OH+
…qui n'ont pas de domaine commun…
La première étape , la plus lente se
produit entre
ces deux espèces, dont le produit des concentrations est maximal
à pH 4
à 5. On remarquera qu'à ce pH les deux espèces
sont largement
minoritaires dans le milieu.
On se place donc à un pH aux alentours de 4-5, qui est le
pH optimum
(c'est un compromis...du genre le moins pire...) . Le mécanisme
est rappelé ci dessous:
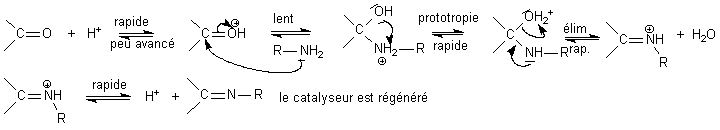
L'imine obtenue est en général
solide et sa
précipitation déplace tous les équilibres
vers la droite de sorte que la réaction atteint tout de
même un bon rendement...
Lors des
actions nucléophiles par un énolate,
l'énolate est obtenu par action de la potasse alcoolique sur
l'aldéhyde ou la cétone utilisée. La base active
est l'alcoolate:
OH
-
+
R-OH
<=> H
2O
+ RO
-
K=10
-2
RO
-
+ R'-CH
2-COH
<=>
ROH
+ R'-CH
--COH
K=10
-4
alcoolate
énolate
On pourra retrouver l'ordre de grandeur des valeurs de pKa
grâce
à l'
échelle proposée en
début de page.
Malgré le faible avancement des deux réactions, on
parvient à réaliser la réaction d'aldolisation
dans ces conditions, par consommation de la base conjuguée par
addition nucléophile sur le carbonyle ou SN sur un
dérivé halogéné.
Toutefois, si l'on souhaite une réaction A/B totale, (voir
échelle de pKa) , on peut utiliser
un
amidure voire un alcanure.
R-NH
-
+ R'-CH
2-COH
<=>
R-NH
2
+ R'-CH
--COH
K=10
+14
Les deux méthodes sont utilisées.
On remarquera que dans le cas de dicétones (pKa=9)
ou
dialdéhydes (pKa= 5) , de la potasse alcoolique suffit amplement
(voire de la soude aqueuse, s'il n'y a pas de soucis particuliers de
solvatation) .
Dans le cas des diesters, l'alcoolate relatif au groupe ester est
imposé pour ne pas induire de saponification (avec la soude ou
potasse par exemple). L'éthanolate est particulièrement
bien indiqué, obtenu par action de Na par exemple.
Les hydrures métalliques LiAlH4
, NaBH4
, LiH ou NaH sont
utilisés en général pour leurs
propriétés nucléophiles, mais ce sont aussi des
bases, conjuguées de H
2 .
Se pose alors le problème du choix du solvant . LiAlH
4
,
LiH , et NaH ne tolèrent pas de solvant protique, c'est à
dire pas de solvant présentant la moindre acidité : seuls
les éthers et les alcanes conviennent.
NaBH
4
présente quant à lui une basicité
inférieure à celle des alcoolates de sorte qu'il
tolère parfaitement les alcools comme solvant.
Même
si basicité et nucléophilie sont des
phénomènes indépendants, on remarquera tout de
même que NaBH4
est aussi le moins nucléophile puisqu'il ne sait pas
réagir avec les esters mais seulement avec les aldéhydes
et cétones.
Si une molécule dont on veut
réduire le groupe C=O présente aussi une fonction acide
carboxylique, il est à retenir que la première
réaction de l'hydrure consistera en une réaction A/B avec
le groupe COOH pour donner COO
- et un dégagement
de
dihydrogène. Tous les protons ayant réagi, alors dans un
deuxième temps, l'excédent (nécessaire) d'hydrure
pourra agir comme nucléophile (en général tout
ceci se produit avec LiAlH
4 )
Donc dans une première étape, se produit une
réaction A/B :
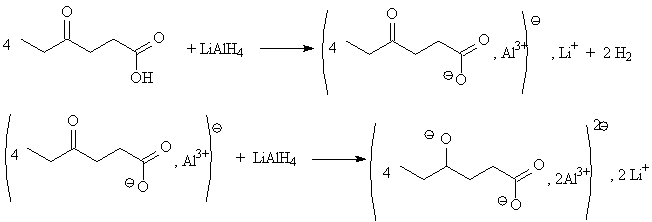
La deuxième étape, a lieu in
situ , mais ne peut
néanmoins avoir lieu que lorsque toutes les fonctions acides
carboxyliques ont été "neutralisées". Il faut donc
mettre l'aluminohydure de lithium en excès (double) pour pouvoir
réaliser la réduction de la fonction cétone. Le
tout est réalisé dans un solvant absolument aprotique
(éther anhydre par exemple).